Talbot helps ID muscle gene that, when altered, causes joint disease
Jared Talbot is part of a 32-member international research team that identified a gene that, when altered, can cause bent fingers and toes, clubfoot, scoliosis, and short stature.
The team discovered that partial loss of the protein coding gene MYLPF (myosin light chain, phosphorylatable, fast skeletal muscle) results in a disorder called distal arthrogryposis (DA) that’s present at birth.
July 23, The American Journal of Human Genetics (AJHG) published the team’s paper “Mutations in MYLPF Cause a Novel Segmental Amyoplasia that Manifests as Distal Arthrogryposis” that details the findings. In May, bioRxiv — a free online and distribution service — also posted a preprint of the paper.
Talbot, an assistant professor in the University of Maine School of Biology and Ecology, is the study’s second author. He contributed equally with Jessica Chong, the first author and an assistant professor of human genetics at the University of Washington.
The discovery has several exciting implications.
“Before a disease can be effectively treated, its cause needs to be understood,” says Talbot. “Right now, DA is treated through surgery, which often has to be repeated several times over a lifetime. By understanding the disease better we may be able to discover longer-lasting and less-invasive ways to treat it.”
More broadly, the breakthrough adds to scientists’ knowledge about how prenatal muscle formation affects health throughout a lifetime, he says.
Chong began the project while working with Dr. Michael J. Bamshad in the University of Washington’s Division of Genetic Medicine. They identified the initial cases and headed an international team that, so far, has identified MYLPF mutations in 19 people with DA in eight families.
Their results also could provide insight into arthrogryposes, which occurs in about 1 in 3,000 births. Arthrogryposes are a larger group of conditions characterized by multiple joint contractures at birth, including in the shoulders, hips and knees. The most common type of arthrogryposis is amyoplasia, which involves muscle loss.
“We hope that our findings may help to shed light on potential genetic causes of amyoplasia, because to date, the etiology of most cases of amyoplasia remains unknown,” says Chong.
Chong and Bamshad also learned of a person with DA who had no muscle in one of their feet, which indicates there may be more similarity than previously thought between DA and amyoplasia.
Bamshad and Chong contacted Talbot, who studies the same gene in zebrafish embryos.
Zebrafish have a similar genetic structure to humans. They share 70% of genes with people and 84% of genes known to be associated with human disease have a zebrafish counterpart. Zebrafish muscle development also mirrors that of people and their embryos grow quickly and are see-through.
Talbot investigated how muscle development is affected by loss of this gene’s function, to understand the “why” behind the human findings. He began work on MYLPF while a postdoc in Dr. Sharon Amacher’s laboratory at The Ohio State University.
There, he mentored Emily Teets, who studied MYLPF function as part of her undergraduate honors thesis. They generated mutations that remove one of the two zebrafish MYLPF genes, called mylpfa, and found that this gene is needed for normal muscle structure and function.
Last fall, Talbot began research at UMaine, where he uses zebrafish to investigate muscle formation and model this human muscle disease.
“MYLPF protein acts in the muscle. We think the crooked joints of DA arise because of reduced muscle function when those joints are forming in the womb,” he says.
“We can’t study people’s muscle strength before they’re born, but we can study zebrafish in their early development and use these fish to model what happens in people.”
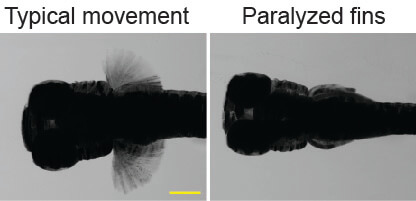
Talbot found that zebrafish with mylpfa knocked out had complete paralysis in their pectoral fin and reduced overall muscle strength.
In addition, he and Teets discovered that muscle eventually degenerated in the zebrafish. This, he says, suggests that some muscle loss that people with DA experience may be due to degeneration in utero.
Talbot used protein models to understand why some of the specific mutations found in humans have dominant inheritance (where one copy of a mutation can cause disease) and other mutations have recessive inheritance, (where two copies must be mutated for a disease to occur).
He found the dominant mutations are caused by changes in parts of the protein that directly contact another protein called myosin, which is the motor protein that contracts muscle.
And, working with David Warshaw at the University of Vermont, they showed that myosin function is reduced in the zebrafish DA model.
Together, these fish and human findings show that MYLPF mutations cause a disease, DA, and they offer insights into how and why that disease arises before birth.
“I have investigated several disease models, but this was the first time that I was able to offer insights from a model organism at the same time that the gene’s function was being connected to a human condition,” says Talbot.
“Our basic-science study paired beautifully with the clinical findings to tell one cohesive story that was enriched by everyone involved.”
Other participating researchers include Samantha Previs, Brit Martin, Kathryn Shively, Colby Marvin, Arthur Aylsworth, Reem Saadeh-Haddad, Ulrich Schatz, Francesca Inzana, Tawfeg Ben-Omran, Fatima Almusafri, Mariam Al-Mulla, Kati Buckingham, Tamar Harel, Hagar Mor-Shaked, Periyasamy Radhakrishnan, Katta Girisha, Shalini Nayak, Anju Shukla, Klaus Dieterich, Julien Faure, John Rendu, Yline Capri, Xenia Latypova, Deborah Nickerson, Paul M. Janssen, and the University of Washington Center for Mendelian Genomics.
Contact: Beth Staples, beth.staples@maine.edu